1. Yeo C.J.J., Darras B.T. Overturning the paradigm of spinal muscular atrophy as just a motor neuron disease. Pediatr. Neurol. 2020; 109: 12-9. DOI: 10.1016/j.pediatrneurol.2020.01.003
2. Deguise M.O., Kothary R. New insights into SMA pathogenesis: immune dysfunction and neuroinflammation. Ann. Clin. Transl. Neurol. 2017; 4(7): 522-30. DOI: 10.1002/acn3.423
3. Wirth B., Karakaya M., Kye M.J., Mendoza-Ferreira N. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu. Rev. Genomics Hum. Genet. 2020; 21: 231-61. DOI: 10.1146/annurev-genom-102319-103602
4. Singh R.N., Howell M.D., Ottesen E.W., Singh N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017; 1860(3): 299-315. DOI: 10.1016/j.bbagrm.2016.12.008
5. Boda B., Mas C., Giudicelli C., Nepote V., Guimiot F., Levacher B., et al. Survival motor neuron SMN1 and SMN2 gene promoters: identical sequences and differential expression in neurons and non-neuronal cells. Eur. J. Hum. Genet. 2004; 12(9): 729-37. DOI: 10.1038/sj.ejhg.5201217
6. Kim J.K., Jha N.N., Awano T., Caine C., Gollapalli K., Welby E., et al. A spinal muscular atrophy modifier implicates the SMN protein in SNARE complex assembly at neuromuscular synapses. Neuron. 2023; 111(9): 1423-39. DOI: 10.1016/j.neuron.2023.02.004
7. Maretina M.A., Zheleznyakova G.Y., Lanko K.M., Egorova A.A., Baranov V.S., Kiselev A.V. Molecular factors involved in spinal muscular atrophy pathways as possible disease-modifying candidates. Curr. Genomics. 2018; 19(5): 339-55. DOI: 10.2174/1389202919666180101154916
8. Chudakova D., Kuzenkova L., Fisenko A., Savostyanov K. In search of spinal muscular atrophy disease modifiers.Int. J. Mol. Sci. 2024; 25(20): 11210. DOI: 10.3390/ijms252011210
9. Crisafulli S., Boccanegra B., Vitturi G., Trifirò G., De Luca A. Pharmacological therapies of spinal muscular atrophy: a narrative review of preclinical, clinical-experimental, and real-world evidence. Brain Sci. 2023; 13(10): 1446. DOI: 10.3390/brainsci13101446
10. Попов К.Д., Алексеева Т.М., Назаров В.Д., Власенко А.И., Малышев С.М. Молекулярные маркеры тяжести заболевания и ответа на терапию нусинерсеном при спинальной мышечной атрофии 5q (обзор литературы). Нервно-мышечные болезни. 2023; 13(3): 33-9. DOI: 10.17650/2222-8721-2023-13-3-33-39
11. Lapp H.S., Freigang M., Hagenacker T., Weiler M., Wurster C.D., Günther R. Biomarkers in 5q-associated spinal muscular atrophy - a narrative review. J. Neurol. 2023; 270(9): 4157-78. DOI: 10.1007/s00415-023-11787-y
12. Фисенко Д.А., Кузенкова Л.М., Куренков А.Л., Увакина Е.В., Попович С.Г. Нейрофиламенты как биомаркер спинальной мышечной атрофии. Неврологический журнал имени Л.О. Бадаляна. 2023; 4(3): 130-6. DOI: 10.46563/2686-8997-2023-4-3-130-136
13. Paris A., Bora P., Parolo S., MacCannell D., Monine M., van der Munnik N., et al. A pediatric quantitative systems pharmacology model of neurofilament trafficking in spinal muscular atrophy treated with the antisense oligonucleotide nusinersen. CPT Pharmacometrics Syst. Pharmacol. 2023; 12(2): 196-206. DOI: 10.1002/psp4.12890
14. Flotats-Bastardas M., Bitzan L., Grell C., Martakis K., Winter B., Zemlin M., et al. Paradoxical increase of neurofilaments in SMA patients treated with onasemnogene abeparvovec-xioi. Front. Neurol. 2023; 14: 1269406. DOI: 10.3389/fneur.2023.1269406
15. Johnson E.W., Sutherland J.J., Meseck E., McElroy C., Chand D.H., Tukov F.F., et al. Neurofilament light chain and dorsal root ganglia injury after adeno-associated virus 9 gene therapy in nonhuman primates. Mol. Ther. Methods Clin. Dev. 2023; 28: 208-19. DOI: 10.1016/j.omtm.2022.12.012
16. Fader K.A., Pardo I.D., Kovi R.C., Somps C.J., Wang H.H., Vaidya V.S., et al. Circulating neurofilament light chain as a promising biomarker of AAV-induced dorsal root ganglia toxicity in nonclinical toxicology species. Mol. Ther. Methods Clin. Dev. 2022; 25: 264-77. DOI: 10.1016/j.omtm.2022.03.017
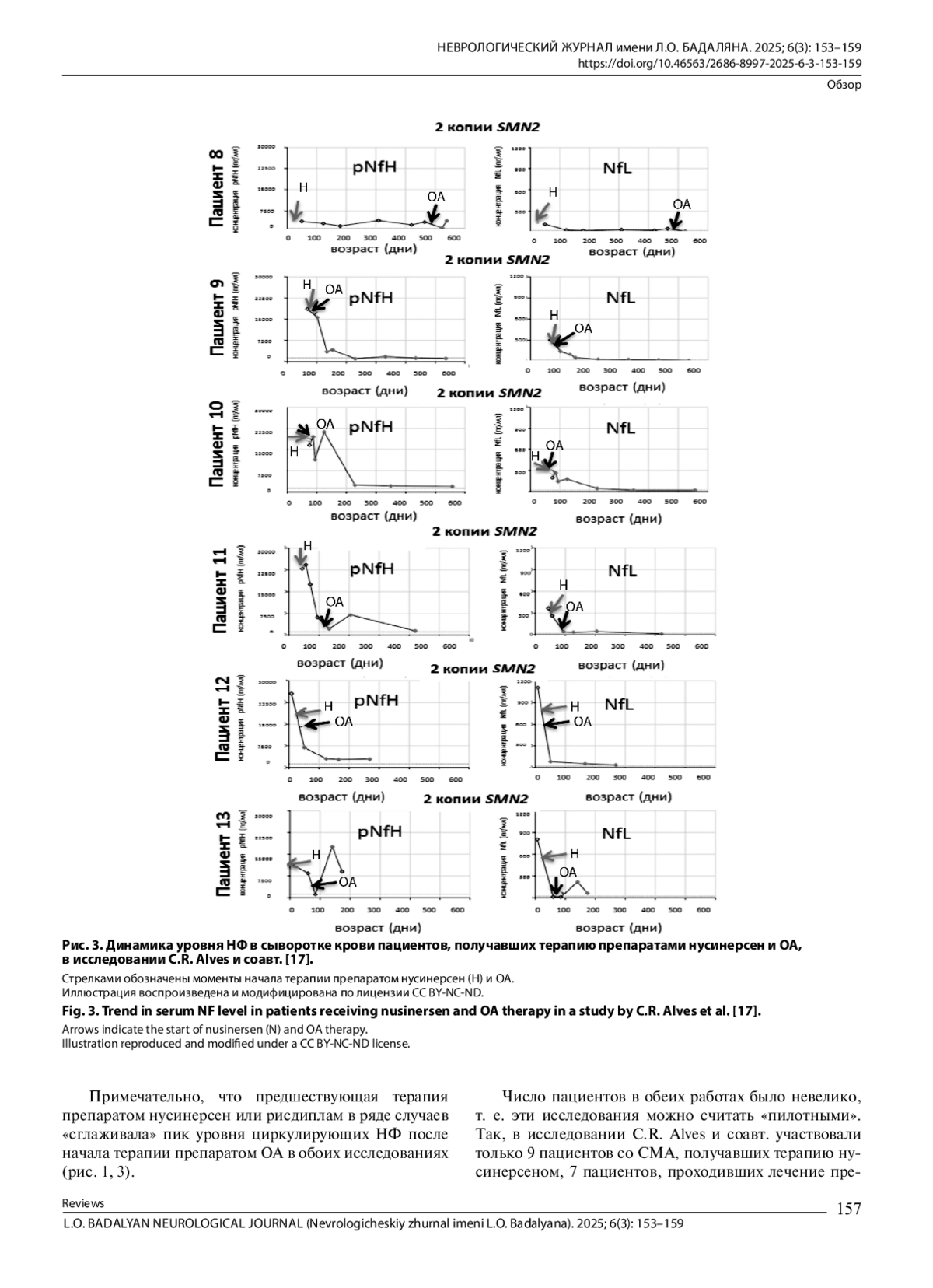
17. Alves C.R., Petrillo M., Spellman R., Garner R., Zhang R., Kiefer M., et al. Implications of circulating neurofilaments for spinal muscular atrophy treatment early in life: a case series. Mol. Ther. Methods Clin. Dev. 2021; 23: 524-38. DOI: 10.1016/j.omtm.2021.10.011
18. Gauthier A., Viel S., Perret M., Brocard G., Casey R., Lombard C. A comparison of Simoa(TM) and Ella(TM) to assess serum neurofilament-light chain in multiple sclerosis. Ann. Clin. Transl. Neurol. 2021; 8(5): 1141-50. DOI: 10.1002/acn3.51355
19. Фисенко Д.А., Кузенкова Л.М., Куренков А.Л., Семикина Е.Л., Увакина Е.В., Черников В.В. и др. Динамика уровней лёгких и тяжёлых цепей нейрофиламентов в сыворотке крови детей со спинальной мышечной атрофией на фоне применения генной терапии. Неврологический журнал имени Л.О. Бадаляна. 2025; 6(1): 26-36.